Cystisk fibrose
Synonymer i en bredere forstand
Cystisk fibrose, lunger
Engelsk: mucoviscidosis, cystisk fibrose
Definition af cystisk fibrose
Cystisk fibrose er en arvelig sygdom. Arven omtales medicinsk som autosomal recessiv. Cystisk fibrose (cystisk fibrose) arves ikke på kønskromosomerne X og Y, men på det autosomale kromosom 7.
Læs vores generelle artikel om metaboliske sygdomme: Metaboliske lidelser - hvad betyder det?
Mutationen er på det såkaldte CFTR-gen. Recessiv betød, at to defekte kopier af genet måtte være til stede for at sygdommen kunne bryde ud. Hvis en person har en sund og muteret genplacering på det tilsvarende kromosom 7, forekommer sygdommen ikke.
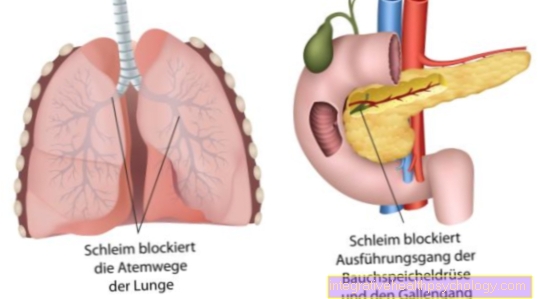
Resultatet er et patologisk genprodukt. Den derved kodede Kloridkanaler er brudt. De defekte chloridkanaler fører til dannelse af tykt slim i alle eksokrine kirtler.
Disse eksokrine kirtler, dvs. kirtler, der frigiver deres sekretion udefra, inkluderer:
- bugspytkirtlen
- tyndtarmen
- luftvejssystemet med lunger og bronkiesystem
- galdekanalen og
- også Svedkirtler
Resumé
Cystisk fibrose er en Arvelig sygdom. Den er arvet på en sådan måde, at den er kønsuafhængig og kun med to defekte gener opstår. Det er mest almindelige autosomale recessive arv.
Konsekvenserne er hårde slimformationer af alle eksokrine kirtler, såsom lungerne, bugspytkirtlen og også svedkirtlerne. De er baseret på det forstyrret transport af klorid mellem cellen indvendigt og udvendigt (læs videre: Klorid i blodet). Det muterede gen er tændt Kromosom 7 og forårsager en bred vifte af organinddragelse med de tilsvarende effekter på vejrtrækning, fordøjelse og reproduktion.
Desværre kan terapi kun lindre symptomerne, men ikke medføre en kur. Det Forventede levealder hos patienter med cystisk fibrose relativt lavt.
Da det er en recessiv arvelig sygdom, er der mennesker, der bærer det ændrede gen, men ikke lider af selve sygdommen. Sådanne personer kaldes Funktionsbærer eller ledere, dvs. bærere. Disse mennesker har ikke cystisk fibrose, fordi den anden kopi af genet er intakt, og den syge ikke er stærk nok til at sejre.
Imidlertid kan hun videregive denne mangelfulde kopi af genet til hendes afkom. Hvis et modificeret gen allerede var tilstrækkeligt til at forårsage en sygdom, ville det være en såkaldt dominerende arv. En sådan arv findes fx i Chorea huntington. Du kan finde ud af mere om denne sygdom under vores emne Chorea huntington.
Ved ca 1:2500 ligger Sygdomsrate hos nyfødte i Tyskland. Carrier handler om alle 25. i den tyske befolkning.
hovedårsagen

Cystisk fibrose er forårsaget af en mutation af et gen på kromosom 7. Dette kromosom er et autosomalt kromosom, ikke et kønskromosom.
Alle har 44 autosomale kromosomer (to identiske versioner af hver) og to kønskromosomer. Denne mutation på kromosom 7 fører til dannelse af defekte chloridkanaler. Reabsorption (re-absorption) af chlorid fra de kirteludskillelser er ikke mulig, fordi receptoren, dockingpunktet for chlorid, ikke er indbygget i kirtelkanalerne.
I stedet sættes det ned til minedrift på grund af det forkerte udseende og struktur. Den naturlige udveksling af chlorid gennem visse chloridkanaler forstyrres. Disse såkaldte kanaler består af proteiner. En lang række proteiner kodes på vores DNA. På grund af den genetiske defekt af kloridkanalerne er der en dehydreret og hård produktion af slim fra alle kirtler, der frigiver deres sekretion udefra. Slimet blokerer derefter delvist kanalerne eller luftvejene i lungerne.
Læs også om dette Kromosommutation
Diagnose af cystisk fibrose

De typiske symptomer, der begynder i spædbarnet, er banebrydende i diagnosen cystisk fibrose.
Denne mistanke styrkes af en positiv familiehistorie (sygdom hos far / mor eller nære slægtninge). En positiv familiehistorie betyder, at der er eller allerede har været tilfælde af cystisk fibrose i familien - på moderlig eller faderlig side.
Manglen på bugspytkirtlenzymer kan også påvises i afføringen. Eventuelle blokeringer i luftvejene kan opdages ved røntgenstråling af brystet.
En svedtest, der måler chloridindholdet i sved, hjælper også med diagnosen cystisk fibrose. Hvis en bestemt værdi overskrides, og de andre symptomer også gælder, er diagnosen relativt fast. Ofte bemærker forældrene selv det øgede saltindhold i spædbarnets sved.
Det ufødte barn kan også testes for denne arvelige sygdom. Ved hjælp af en fostervandsprikker (fostervandsprøve) føtalceller fjernes og undersøges for det muterede gen.
Læs mere om emnet: Røntgenundersøgelse af barnet
Terapi af cystisk fibrose
Enhver, der er berørt af cystisk fibrose, vil modtage rådgivning i en Cystisk fibrose - ambulant afdeling eller råd fra Human genetiker (Specialist i arvelige sygdomme) anbefales. Disse kan hjælpe med at øge livskvaliteten eller, hvis du vil have børn, beregne sandsynligheden for et sygt barn. Forudsat at forældrene er frugtbare og frugtbare.
Ellers er behandling symptomatisk, da årsagen, det defekte gen, ikke kan elimineres.
Uhelbredelig sygdom
Cystisk fibrose (cystisk fibrose) er stadig en uhelbredelig sygdom i dag.
I tilfælde af cystisk fibrose er det vigtigt at have et tilstrækkeligt indtag af bordsalt (Natriumchlorid, NaCl). Mucolyse er selvfølgelig rettet mod. Mukolyse er opløsning af slim, især i lungerne, for at gøre vejrtrækningen lettere.
Medicin og indånding kan lindre symptomerne. Hvis lungefunktionen mærkbart forværres, kan der gives ilt.
Gennem intensiv fysioterapi (fysioterapi), for eksempel at tappe massage og åndedrætsøvelser, behandles lungeforandringer forårsaget af cystisk fibrose også.
Ofte ender sygdommen med en krævet lungetransplantation. Ventelisterne er dog lange.
Oral indgivelse af pancreas-enzymer og fedtopløselige vitaminer er også en del af behandlingen. Derfor skal bugspytkirtelens opgave understøttes eller snarere erstattes. Fedtopløselige vitaminer er A, D, E og K. De skal gives direkte i blodet, da de ikke kan optages fra mad på grund af mangel på fordøjelsesenzymer.
Diæten bør også indeholde mange kalorier, da kun en brøkdel af dem kan fås fra mad.
For at undgå yderligere risikofaktorer for komplikationer som influenza eller lungebetændelse, bør barnet vaccineres. Følgende vaccinationer anbefales:
- mæslinger
- pneumokokker
- influenza
Læs mere om emnet: superinfektion
Naturligvis kræver disse foranstaltninger en konsultation med en læge, som risiciene bør drøftes med.
I dag er der et stort håb for cystisk fibroseterapi i genetisk forskning. Man forsøger at introducere den manglende genetiske information i det menneskelige genom. Vi leder efter vektorer, der kan mestre denne opgave. Vektorer kan for eksempel være bakterielt eller viralt DNA, der formår at inkorporere den sunde frekvens i vores genetiske sammensætning.
Den terapeutiske tilgang hos ufødte patienter testes i øjeblikket. Hos mus er det allerede lykkedes med musembryoerne at introducere det raske gen, der indeholdt den rigtige gensekvens, gennem fostervand (amniotisk væskeinokulation). Det sunde CFTR-gen blev således produceret i disse mus. Amniocentese er en punktering og fjernelse af barnets celler fra fostervandet, hvilket gøres gennem moderens mavevæg.
I Tyskland er denne form for intrauterin (= i livmoderen = i livmoderen) "terapi" imidlertid forbudt.
profylakse
EN forebyggende foranstaltning i denne forstand findes det ikke, fordi det er en arvelig sygdom.
Imidlertid kan et humant genetisk rådgivningscenter (normalt findes på universitetshospitaler) besøges. Her beregnes det, hvor stor risikoen ville være at overføre sygdommen til børn.
Dette råd er altid nyttigt, hvis der er en familiehistorie med cystisk fibrose.
Også en Prenatal diagnostik er værd at stræbe efter. Her før fødselen (dvs. prenatalt) a Fostervandsundersøgelse (fostervandsprøve) udført. Fosterceller (celler fra barnet) er taget fra fostervandet, og DNA'et undersøges for det muterede gen.
Prognose af cystisk fibrose
Desværre er den gennemsnitlige levealder hos patienter med cystisk fibrose kun 32-37 år. I dag estimeres levealderen for nyfødte, der er født med denne tilstand, til at være omkring 45-50 år.
Prognosen afhænger meget af behandlingen, og om den overholdes.
Patienten selv og hans motivation spiller derfor en vigtig rolle.