hæmofili
Synonymer
Hæmofili, arvelig blødningsforstyrrelse, mangel på blodkoagulationsfaktor,
Faktor VIII-mangel, faktor IX-mangel, hæmofili
definition
Hæmofili er en arvelig sygdom i blodkoagulationssystemet:
De berørte patienter har nedsat blodkoagulation, hvilket manifesterer sig i, at blødning opstår med det mindste traume, og blødningen er vanskelig at stoppe.
Koagulationsfaktorer kan ikke aktiveres, så det regelmæssige forløb i koagulationskaskaden ikke er muligt.
Hæmofili er en medfødt sygdom, der findes i to former,
Hæmofili A og B kan være til stede.
Hæmofili A forekommer hyppigere (i 85% af tilfældene) end form B (ca. 15%) og er den mere alvorlige af de to former.
Aktiviteten af faktor VIII-komplekset er begrænset i hæmofili A, fordi dannelsen af faktor VIII-C i levercellerne er reduceret.
Faktor VIII-C er en af de to underenheder i faktor VIII-komplekset: von-Willebrand-Faktor og faktor VIII-C danner dette koagulationsfremmende kompleks i koagulationskaskaden. Von Willebrand-faktoren beskytter faktor VIII-C mod at nedbrydes for hurtigt.
Mængden af funktionel faktor VIII-kompleks reduceres følgelig i form A.
I hæmofili B. syntese af koagulationsfaktor IX finder sted i mindre grad, så dens virkning i koagulationsprocessen er fraværende og hæmostase nedsættes.
Forekomst / epidemiologi
Forekomsten af hæmofili i befolkningen er for hæmofili A. 1 ud af 5.000 og til form B. 1:15.000.
Det faktum, at især mænd bliver syge, skyldes kønsrelaterede problemer
X-forbundet form for arv af koagulationsdefekten (se årsag / udvikling).
Grundlæggende
Blodkoagulation er et fint afstemt system: Forskellige komponenter i blodet, de såkaldte koagulationsfaktorer, aktiveres efter hinanden på en kaskadelignende måde for at muliggøre hæmostase i tilfælde af en skade.
Koagulationssystemet består af primær og sekundær hæmostase, der også kaldes plasmatisk koagulering.
Hvis en karvæg er skadet, indsnævres det berørte fartøj i et første trin (= primær koagulation). Så en rekvisitter (=Trombocyttromben) ved det skadede område, der er dækket af blodplader (= Blodplader) dannes for at opnå en initial forsegling af fartøjet.
Dannelsen af tromben aktiverer den plasmastiske koagulation:
Koagulationskaskaden starter, og samspillet mellem de forskellige koagulationsfaktorer skaber et stabilt sår eller vaskulær lukning.
Årsag og oprindelse
Det hæmofili Begge former for hæmofili arves som en X-bundet recessiv egenskab.
I den såkaldte X-bundne tilstand af arv af hæmofili ligger den genetiske information for et genprodukt på kønskromosomet X, som findes i dobbelttal hos kvinder og i enkelttal hos mænd.
Der er to gener til enhver genetisk information, de såkaldte alleler. Hvis der er recessiv arv, skal begge alleler påvirkes af en mutation for at forårsage sygdommen: recessiv betyder, at den patologisk ændrede genetiske information stammer fra en sund, dvs. ændres ikke, information undertrykkes og fører ikke til sygdom. En recessiv arvelig sygdom udvikles kun, hvis begge alleler bærer forkerte genetiske oplysninger.
Er kun -en Genudtryk ændret sig, kom det Ikke til hæmofili.
Info: arv
Hvis kun en genekspression ændres, vil der ikke være nogen sygdom.
Da mænd kun har et X-kromosom i kønkromosomparet, og derfor er information om genprodukter på deres X-kromosom kun tilgængelig i en enkel form, vil de blive syge, hvis kun en allel er defekt.
På den anden side kvinder, der har to X-kromosomer som kønskromosomer, bliver kun syge, når begge kromosomer har defekte gener for genproduktet.
Hos mænd, der lider af hæmofili, er allelen, der bærer den arvelige information for koagulationsfaktor VIII-C (hæmofili A) eller koagulationsfaktor IX (hæmofili B), kun tilgængelig i en let modificeret form;
Kvinder er nødt til at bære denne genetiske ændring på begge alleler for at være syge.
Hvis kvinder har en defekt og en ikke-ændret allel, kaldes de bærere af en X-bundet arvelig sygdom, såsom hæmofili.
Ved hæmofili har den defekte genetiske information effekten af at reducere aktiviteten af den påvirkede koagulationsfaktor eller at reducere produktionen af koagulationsfaktor.
X-kromosomet er bæreren af det genetiske materiale til koagulationsfaktorer for sekundær hæmostase.
Ca.. 70% af mutationerne i det genetiske materiale, der forårsager hæmofili, overføres i familier, 30% af sygdommene skyldes spontane ændringer (= spontane mutationer) i den genetiske information.
diagnose
Efter at have spurgt om patientens medicinske historie og foretaget en grundig fysisk undersøgelse, er der yderligere trin i diagnosen hæmofili:
I 2/3 af tilfældene er der tilfælde af hæmofili i familien, hvorfor der bør stilles en familiel klynge af sygdommen, om patienten giver lægen symptomer, der er mistænkelige for hæmofili.
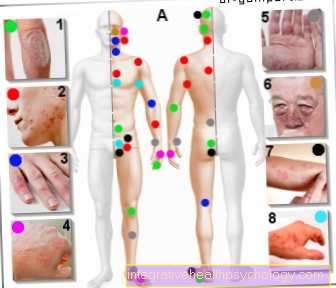
Patienterne rapporterer om blå mærker som et resultat af de mindste skader.
Man skelner dem Alvorlighed af hæmofili ind i en mild, moderat eller svær form for sygdommen.
Undersøgelse af en blodprøve giver følgende resultater, der er typiske for hæmofili:
Blødningstiden er normal (= den primære koagulering er intakt), men funktionen af den plasmatiske koagulering reduceres, hvorfor den såkaldte PTT-tid er længere.
PTT står for den delvise thromboplastintid. Det måles i laboratorieundersøgelser for at kontrollere funktionen af faktorer I, II, V, VIII til XII samt XIV og XV.
Da en faktor i koagulationskaskaden mangler ved hæmofili, kan den ikke køre optimalt, hvilket resulterer i en forlænget koaguleringstid.
For at være i stand til at skelne mellem hæmofili A og B, skal patientens blod undersøges for faktorer VIII og IX; fraværet af de to faktorer bestemmer typen af hæmofili.
Differential diagnose
Hæmofili skal differentieres fra andre lidelser i koagulationssystemet, idet der især nævnes von Willebrand-Jürgens syndrom.
Von Willebrand-faktoren (vWF) fungerer sammen med faktoren VIII-C im
Faktor VIII-kompleks og forårsager hæmning af den for tidlige nedbrydning af faktor VIII, derudover fremmer faktoren koagulation ved at mediere vedhæftningen af blodplader til det sårede vaskulære sted. Således er vWF en vigtig del af både primær og sekundær koagulering.
Hvis von Willebrand-Jürgens syndrom er til stede, er dannelsen af
vWF'er i fartøjets væggceller finder kun sted i mindre omfang eller på en forkert måde. Den reducerede uddannelsesgrad eller den utilstrækkelige funktion af
von Willebrand-faktor er forårsaget af mutationer i genet til faktordannelse. Arven fra syndromet eller den årsagsmæssige genmutation arves på en autosomal dominerende måde: den genetiske information for vWF er på kromosom nummer 12, som ikke er et kønskromosom. I den dominerende arvsmåde undertrykker en syg allel virkningen af den anden, sunde allel, så sygdommen er forårsaget af mutationen af et gen og forårsager kliniske symptomer.
Patienterne lider typisk af hud- og slimhindeblødning, mindre af spontan blødning som hæmofili-patienter.
Sygdommen bemærkes ofte kun, når der er langvarig blødning efter en operation.
Patientens blødningstid efter en skade forlænges, og værdierne for den plasmatiske koagulation ændres patologisk (forlænget PTT).
Von Willebrand-Jürgens syndrom er forårsaget af indgivelse af desmopressin eller udskiftning af faktor VIII og vWF for at stabilisere koaguleringen (for en mere detaljeret forklaring se under "Terapi med hæmofili").
Hvad er symptomerne på hæmofili?
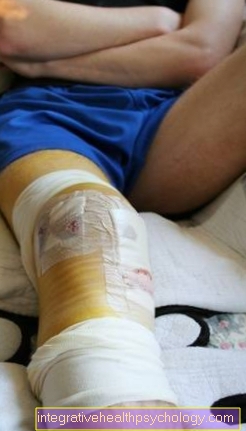
Symptomerne i form af hæmofili adskiller sig ikke:
- Hæmofili fører normalt kun til kliniske symptomer hos mænd.
- Overdreven blødning forekommer med hæmofili, som ikke er i forhold til den tidligere, for det meste banale ulykke (trauma), er blødningstiden normal, men blødning forekommer typisk, der ikke forekommer hos raske mennesker.
- Der kan sondres mellem tre former for hæmofili, som er defineret af den resterende aktivitet af de berørte koagulationsfaktorer:
1.svær hæmofili (hæmofili) med mindre end 1% restaktivitet eller en andel af den funktionelle faktor, der stadig er til stede, hvor spontan blødning forekommer og ledblødning forekommer
2. Moderat hæmofili (blodsygdom) med faktoraktivitet mellem
1 og 5% af den normale faktoraktivitet og forekomsten af blå mærker (= hæmatomer) efter mindre traumer
3. mild hæmofili (hæmofili) hvori der stadig er 5-15% restaktivitet, og hvor patienter rapporterer symptomer såsom hæmatomer (blå mærker) efter betydelig traume og postoperativ blødning.
Læs mere om emnet: Hvad er årsagerne til en hjerneblødning
- Kvinder, der normalt kun har ændret genetisk information for hæmofili, viser normalt ingen kliniske symptomer med over 50% restaktivitet.
- Patienter oplever blødning i store led som sådan knæ-, Hofte- eller Skulderled, hvor man taler om hæmarthrose. Blødningen udløser en betændelsesreaktion med reparationsprocesser i leddet, hvilket kan føre til en afstivning af leddet.
- Det kan også blø i muskulatur og det bløde væv kommer. Blødningen fører til en stigning i trykket i musklerne eller vævet, da der nu er mere volumen. Dette kan især påvirke dine arme og ben Rummets syndrom at lede:
Det øgede tryk forårsager komprimering af blodkar og nerver, så ekstremiteterne underforsynes og store områder af væv kan dø. Rumssyndrom skal behandles af kirurgen så hurtigt som muligt for at forhindre tab af ekstremiteten. - Blødning ind i maven opstår, hvilket er en livstruende situation for patienten.
- Ualmindelig langvarig blødning er mulig efter operationen.
Derudover kan der være lange perioder med blod i urinen. Her truer man Anæmifordi patienten konstant mister blod, muligvis ubemærket. - Er særlig farlige Hjerneblødning (= intrakraniel blødning), hvilket fører til død hos 10% af mennesker med hæmofili. Du kan finde ud af mere om dette emne under vores emne Hjerneblødning.
Hvordan udføres terapi mod hæmofili?

Blødning skal forekomme hos de syge hæmofili skal undgås, hvorfor patienten ikke gør det Medicin der hæmmer blodkoagulation såsom Acetylsalicylsyre (Aspirin®) og ingen intramuskulære injektioner (= sprøjter i muskulatur) henholdsvis.
Hvis der opstår et traume med blødning, er omhyggelig lokal hæmostase af stor betydning for at forhindre blødning i nabovævet.
Lægemiddelterapi af hæmofili består i udskiftning af koagulationsfaktorer, som kroppen ikke kan producere i tilstrækkelige mængder.
Patienter med let hæmofili modtage koagulationsfaktorpræparaterne efter behov, dvs. hvis der har været traumer med blødning, eller hvis der er planlagt større operationer, hvilket kan resultere i rigelig blødning.
Profylaktisk faktorudskiftning bør anvendes til patienter med moderat til svær hæmofili, dvs. den manglende faktor skal gives permanent Før blødning forekommer.
Hvis der er restaktivitet af faktor VIII på mere end 15% eller faktor IX på mere end 20-25%, er regelmæssig behandling ikke nødvendig; disse patienter får den manglende koagulationsfaktor i tilfælde af spontan blødning eller før planlagt operation.
Langvarig terapi såvel som før operationen kan tage form af hjemme-selvbehandling, hvor patienten selv anvender den manglende koagulationsfaktor.
Der er et behandlingsalternativ for patienter med let hæmofili:
Det aktive stof Desmopressin (f.eks. Minirin®) fører til en fordeling af
Faktor VIII, der er opbevaret i fartøjets vægge.
Imidlertid kan lægemidlet kun gives i et par dage ad gangen, da hukommelsen i karvæggene efter stimulering af faktorfrigivelsen er opbrugt og skal gentages igen.
I akut terapi hæmofili der er tre muligheder for intervention:
- Patienten modtager aktiveret protrombin, et stof, der fremmer blodkoagulation, så blodet stoppes så hurtigt som muligt.
- En anden terapeutisk mulighed er administration af faktor VII-præparater. Denne faktor er i begyndelsen af koagulationskaskaden og starter dets regelmæssige forløb.
- Den tredje terapeutiske mulighed er anvendelsen af dyrefaktor VIII-C for at gøre det muligt for patienten at stoppe blødning.
Komplikationer
Erstatningsgaven (=substitution) af koagulationsfaktorer kan føre til dannelse af antistoffer mod nøjagtigt disse faktorer, så substitutionen i en konstant dosis ikke længere har nogen terapeutiske virkninger, hvorfor man taler om dannelsen af hæmmere.
Afhængig af bestemmelsen af koncentrationen af antistofferne i patientens blod kan høje doser af faktor VIII administreres med det formål at genvinde tolerance over for denne faktor og eliminere dannelsen af antistoffer. Denne procedure skal kun udføres i specialiserede centre.
Der er en lav risiko for at få hepatitis eller få HIV-virus (=HIV) da faktorerne stammer fra humane blodprodukter.
En anden risiko for substitutionsterapi med hæmofili er forekomsten af allergiske reaktioner.